Health Today
Inflammation, Stress and Metabolic Diseases


Studies in our lab and others have clearly demonstrated that chronic inflammation is a central feature of obesity and the associated metabolic disease cluster. This inflammatory response is distinct, appears to respond to intrinsic cues, and does not resemble the classical inflammatory paradigm. New names have been suggested to describe this phenomenon including “metaflammation” or “paraflammation”.
We examine the molecular mechanisms leading to the emergence of these inflammatory responses and how they are linked to metabolic homeostasis as well as disease. Our effort is targeted to major cell types and organs where inflammatory and metabolic pathways interface, such as adipose and liver tissue as well as macrophages. In these systems and various genetic models, we explore the hormonal and metabolic signals that generate profound effects on systemic endocrine equilibrium.
Obesity-related activation of the serine/threonine kinases, such as JNK, and the consequent inhibition of insulin receptor signaling via phosphorylation of a substrate of insulin receptor, IRS-1 is a central mechanism of insulin resistance. In mice lacking JNK genes, there is dramatic protection from obesity and diabetes. There is also genetic evidence that JNK activation is linked to type 2 diabetes in humans. Currently, we are investigating the detailed molecular mechanisms, target cell types and organs and different JNK isoforms underlying this crosstalk. We also investigate the metabolic signals and stresses that give rise to JNK activation and explore therapeutic and preventive possibilities for diabetes, obesity, and atherosclerosis by blocking JNK function. The ability of nutrients to trigger inflammation raises an important question regarding the control of overt inflammation during physiological fluctuations in nutrient and energy exposure. In search for molecules that prevent such aberrant responses, we recently identified a new class of molecules called STAMP that control nutrient-induced inflammatory responses, particularly in adipocytes. These molecules are nutritionally regulated, particularly in visceral adipose tissue, and their absence results in visceral adipose tissue inflammation, stress responses, and insulin resistance under regular dietary conditions. We are currently investigating the molecular mechanisms of actions of these molecules and studying their target cells and organs.
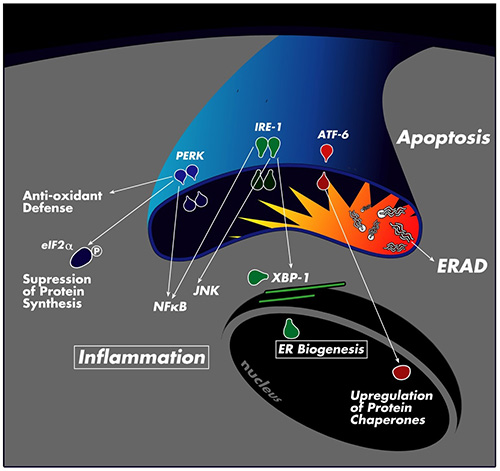
We are also broadly pursuing the molecular mechanisms of the crosstalk between inflammatory and metabolic pathways or integration of nutrient and pathogen sensing pathways. These studies have recently led us to the discovery of endoplasmic reticulum (ER) stress as a central mechanism linking metabolic stress with insulin resistance and type 2 diabetes. ER is a critical organelle responsible for the synthesis, maturation, folding and transport of all secreted and transmembrane proteins. It is also the site for lipid synthesis and packaging. ER meets the fluctuations in its functional capacity by mounting an adaptive response called “unfolded protein response” or UPR. This system helps adapt ER folding capacity and manages ER stress by regulation of protein synthesis and breakdown and also by activating a transcriptional program to assist ER with the necessary components to establish equilibrium. In addition to proteins, ER is also exquisitely sensitive to energy status, nutrients, and pathogens, hence it can integrate these pathways that are central to metabolic pathways.
Obesity also leads to ER stress in metabolically sensitive tissues such as adipose and liver tissues and pancreatic islets. Through activation of JNK and other stress signaling pathways, ER is linked with regulation of insulin action and glucose and lipid metabolism. Currently, we are exploring the molecular mechanisms leading to ER stress in obesity and investigating the role of different UPR branches in metabolic homeostasis. We are also developing strategies for chemically and genetically targeting these pathways for novel therapeutic opportunities against metabolic diseases.
View Source Article Stress, depression may affect cancer survival
Stress, depression may affect cancer survival Defending Against Disease With an Anti-Inflammation Lifestyle
Defending Against Disease With an Anti-Inflammation Lifestyle Increased cardiovascular risk in HIV-infected patients may relate to arterial inflammation
Increased cardiovascular risk in HIV-infected patients may relate to arterial inflammation Inflammation Gone Wild Can Lead To Macular Degeneration
Inflammation Gone Wild Can Lead To Macular Degeneration Chronic Inflammation and Late-Life Decline
Chronic Inflammation and Late-Life Decline INFLAMACIÓN CRÓNICA: EL PROBLEMA DE SALUD DEL CUAL NO HAS ESCUCHADO
INFLAMACIÓN CRÓNICA: EL PROBLEMA DE SALUD DEL CUAL NO HAS ESCUCHADO Dwelling On Stressful Events Can Increase Inflammation in the Body, Study Finds
Dwelling On Stressful Events Can Increase Inflammation in the Body, Study Finds Why Cancer and Inflammation?
Why Cancer and Inflammation? Chronic Inflammation: Reduce It to Protect Your Health
Chronic Inflammation: Reduce It to Protect Your Health Inflammation, Stress and Metabolic Diseases
Inflammation, Stress and Metabolic Diseases Researchers discover link between inflammation and spread of breast cancer
Researchers discover link between inflammation and spread of breast cancer The New Science Behind America's Deadliest Diseases
The New Science Behind America's Deadliest Diseases Foods That Fight Inflammation — And Why You Need Them
Foods That Fight Inflammation — And Why You Need Them MIT confirms link between inflammation, cancer
MIT confirms link between inflammation, cancer Is Chronic Inflammation the Key to Unlocking the Mysteries of Cancer?
Is Chronic Inflammation the Key to Unlocking the Mysteries of Cancer? What Causes Chronic Inflammation?
What Causes Chronic Inflammation? Chronic Inflammation: Reduce It to Protect Your Health
Chronic Inflammation: Reduce It to Protect Your Health Markers of Inflammation and Cardiovascular Disease
Markers of Inflammation and Cardiovascular Disease FAMILY HEALTH GUIDE
FAMILY HEALTH GUIDE The Anti-Inflammation Diet
The Anti-Inflammation Diet